This site uses cookies!
We use cookies for security, analytics and session persistence reasons. Please refer to our Cookie Policy for more information.
General Information
On this page a more detailed explanation of the data present in OLIDA and the different ways to access this data is detailed. This documentation introduces the different concepts and entities that are part of OLIDA and describes the features of this web interface in detail. An overview of the capabilities of the REST API is provided in addition to the descriptions of the data structure. Finally, tutorials illustrate the core use-cases for OLIDA and the F.A.Q. section answers some of the most frequently asked questions.
- Genetic evidence - Familial evidence: This score represents the strength of familial evidence that the variant combination is linked to by looking at the segregation of variants in the pedigree described in the corresponding publication according to the related phenotype and both the patient(s) and the healthy individuals. Since this information could only be obtained from the original publication, this type of evidence only has one associated score: FAMmanual.
- Genetic evidence - Statistical evidence : Statistical evidence that a variant combination does not occur by chance in an individual with the observed phenotype can be obtained using either a) a control cohort of matched ethnicity and sufficient size, or b) one (or more) of the numerous available databases containing genetic data of individuals. The statistical evidence is scored based on the information present in the article (STATmanual) and by checking the presence of the variant combination in the 1000 Genomes project and the citations linked with the involved variants in Clinvar, to assess their link with the studied disease (STATknowledge). The STATmeta score then combines these two scores by taking the maximum of the two values. However, when the variant combination is found in an individual of the 1000 genomes project, the STATmeta takes the value of 0.
- Functional evidence - Gene combination evidence: The evidence for the functional relationship between the involved genes of an oligogenic combination (e.g. biological processes, co-expression, protein-protein interactions) and their relevance for the studied disease phenotype (e.g. experiments on animal models, pathways linked with phenotype), which can be obtained through experiments in vivo, in vitro or in silico with computational analyses. The gene combination evidence score is based on the information present in the article (GENEmanual) and on the protein-protein interaction (PPI) and pathway information from the comPPI, KEGG and Reactome databases (GENEknowledge). The GENEmeta score then combines these scores by taking the maximum of the two values.
- Functional evidence - Variant combination evidence: The evidence for the joint effect of the variants of an oligogenic combination on the observed phenotype, in vivo (e.g. animal models), in vitro (e.g. cell cultures) and/or in silico (e.g. pathogenic effect predictors). The variant combination evidence score is based on the information present in the article (VARmanual) and by using variant pathogenicity information from different pathogenicity predictors: SIFT, MutationTaster2, CADD and Polyphen2 (VARknowledge). The VARmeta score then combines these scores by taking the maximum of the two values.
- Functional score : This score represents the joint gene and variant functional evidence, based on the article information only (FUNmanual) or on the article and databases information (FUNmeta). It is obtained by combining the gene combination and variant combination scores using a decision tree.
- Final score : This score represents the overall confidence of the pathogenicity of the oligogenic combination and its link to the observed phenotype, based on the article information only (FINALmanual) or on the combination of the article and databases information (FINALmeta). It is obtained by combining the familial score, statistical score and functional score with the help of a decision tree.
- Synvar: The genomic coordinates for most variants were obtained using Synvar, a tool which uses text mining to generate many possible synonyms of variant descriptions.
- Varsome: Variants for which the genomic coordinates could not be obtained in Synvar were searched manually in Varsome, a genetic variation search engine.
- Ensembl VEP: A large part of the variant information was retrieved using the Ensembl VEP tool. The variant effect, SIFT and Polyphen2 predictions as well as all the allele frequencies of the variants in large databases were obtained using this tool.
- Crossref: The automated retrieval of data on publications that were added to OLIDA is achieved by using the Crossref API.
- OpenCitations: Publications that cite OLIDA or DIDA, are kept up to date with the help of the OpenCitations API. This allows for entries to be kept up to date with minimal human effort.
- Recommended: to the GitHub repository that was created for this purpose. The README on the front page of this repository details how to submit feedback in the Submitting bugs, feedback and feature requests section.
- Directly contact the developers at olida@ibsquare.be.
Table descriptions
- OLIDA ID – The oligogenic variant combination identifier in OLIDA.
- OMIM Idspecific - IDs in The Online Mendelian Inheritance in Man (OMIM) that pertain to disease phenotypes that have been linked to this specific combination of genetic variants.
- Oligogenic effect – Digenic instances in OLIDA are categorized into one of three classes: True digenic instances refer to combinations where the variants are all mandatory for the appearance of the disease. Monogenic + modifier instances refer to combinations where a variant in one gene is enough to develop the disease but carrying a second one on another gene affects the severity of the phenotype or age of onset. Dual-Molecular diagnosis cases refer to combinations where each variant causes a disease by itself and both phentoypes are present if the combination of variants is present. Oligogenic variant combinations of more than two genes can not be categorized in these classes and are simpy labeled oligogenic.
- Diseases – Orphanet is the reference portal for information on rare diseases and orphan drugs, for all audiences. Orphanet’s aim is to help improve the diagnosis, care and treatment of patients with rare diseases. This column represents the name of the disease as present in Orphanet. Clicking a name will bring you to the detail page on this disease. If a patient’s phenotype could not be associated to a particular orphanet disease, the name of the disease is the one from the article description.
- Ethnicity – When available this column indicates the ethnicity of the population(s) where this combination was found.
- References – This column displays the pubmed ID of the publication where the oligogenic combination and associated evidence was described. If several publications discuss the same combination, their respective PMIDs will be listed here. Clicking on an ID will bring you to the detail page for that article.
- Associated variants – The genetic variants that make up this combination. The table contained in this column lists all variants with their database id, name of the gene in which the variation occurs, the change in coding DNA of the allele and the resulting protein change.
- FAMmanual - Quality of the segregation of variants with the observed phenotype within the family, using both healthy individuals and patients, found in the publication.
- STAT - Scores quantifying the quality of the statistical evidence for the combination that supports the involvement of the combination in the disease, with respect to the control group size and the ethnicity.
- STATmanual - Quality of the statistical evidence based on article information only.
- STATknowledge - Quality of the statistical evidence based on the presence/absence of the combination in external databases.
- STATmeta - Quality of the statistical evidence based on both article and external database information.
- GENE - Scores quantifying the quality of the evidence supporting the functional involvement of the genes in the disease.
- GENEmanual - Gene functional evidence based on the article information.
- GENEmanual_harmonized - Gene functional evidence based on the article information harmonized based on the articles of the database that describe the same gene combination. The maximum score is kept.
- GENEknowledge - Gene functional evidence based on external databases.
- GENEmeta - Maximum of the manual and knowledge scores for the gene evidence, across all the publications.
- VAR - Scores quantifying the evidence supporting the functional involvement of the variants in the disease.
- VARmanual - Variant functional evidence based on the article information.
- VARknowledge - Variant functional evidence based on external databases.
- VARmeta - Maximum of the manual and knowledge scores for the variant evidence.
- FUN - Scores quantifying the evidence supporting the functional involvement of genes and variants in the disease.
- FUNmanual - Functional evidence based on the article information only.
- FUNmeta - Functional evidence based on article and external databases information.
- FINAL - Overall confidence score of the involvement of the oligogenic variant combination in disease.
- FINALmanual - Final score based on article information only.
- FINALmeta - Final score based on article and external databases information.
NOTE: Depending on variant type, certain columns may or may not be present in the table. We discuss all columns of different tables and will indicate when a column is only present in a specific variant table.
- DB ID – The variant identifier in OLIDA.
- Genomic position Hg19 – This value represents the position of the variant-of-interest in the human genome version Hg19/GRCh37.
- Genomic position Hg38 – This value represents the position of the variant-of-interest in the human genome version Hg38/GRCh38.
- Variant effect – This represents the effect of the variant-of-interest as computed by the Ensembl VEP.
- Chromosome – The chromosome on which the genetic variation is located.
- Gene Name – Name of the gene in which the variation has occurred.
- Ref Allele – [Small variants only! This represents the nucleotide(s) that is (are) present in the human reference genome at the position of the variant-of-interest.
- Alt Allele – [Small variants only! This represents the new nucleotide(s) that is (are) present at the position of the variant-of-interest.
- cDNA change – [Small variants only! This represents the change at the c(oding)DNA level for the variant-of-interest. Example: c.1022C>A represents a change in the cDNA at position 1022, where the reference nucleotide is a ‘C’ that changes to an ‘A’. These values are encoded according to the HGVS sequence variant nomenclature.
- Protein change – [Small variants only! ] This represents the change at the amino acid level for the variant-of-interest. Example: p.(A341E) represents a change in the protein sequence at position 341 where the reference amino acid ‘A (Alanine)’ changes to ‘E (Glutamic acid)’. These values are encoded according to the HGVS sequence variant nomenclature.
- Transcript Id – This represents the unique NCBI transcript ID for the gene-of-interest. The cDNA change and protein change values are based on the transcript referenced in this column.
- Location – [CNV variants only! ] This represents the location where the CNV occurs.
- Sequence – [CNV variants only! ] This represents the sequence that is deleted, duplicated or repeated.
- Number of repeats – [CNV repeats variants only! ] The numbers in this column represent the lower bound and the upper bound of the number of repeats of the repeated sequence.
- Type – [CNV variants only! ] The type of CNV: duplication, deletion or repeat.
- Dbsnp ID – [SNP variants only! ] The dbSNP database serves as a central repository for both single base nucleotide subsitutions and short deletion and insertion polymorphisms. The column reports the unique variant ID for dbSNP version 141.
- Variant Type – [InDel variants only! ] Indicates whether the variant is an insertion or a deletion.
- Flag - This flag represents the way the genomic coordinates of the variant were obtained and eventual anomalies in this process. The information of the variants present in the article typically used the gene and cDNA and/or protein change in HGVS format. In order to obtain the genomic coordinates of these variants, the Synvar software was used. The variants that could not be mapped by the software, they were manually searched for using VarSome or dbSNP. The following flags were assigned:
- automatically_attributed - if the genomic coordinates were obtained using Synvar.
- automatically_attributed_and_verified - if the variant was annotated by Synvar and was manually confirmed
- manually_attributed - if the coordinates could not be obtained using Synvar, but were found in a database (using Varsome, dbSNP or).
- manually_corrected - if the curators had to manually correct either the cDNA or the protein change, due to incorrect or expired information from the relevant paper, and the resulting variant was found in a database.
- ambiguous_variant - if the variant found in the database needed a correction in both the cDNA change and the protein change, making it impossible to determine which variant the authors of the article were actually referring to.
- missing_from_databases - if based on the information contained in the article, the variant could not be found in any databases.
- Dataset Allele frequency – [Small variants only! Frequencies of this variant in certain datasets of genetic
data on a population.
- 1000 Genomes Project – The 1000 Genomes Project aims to provide a deep characterization of human genome sequence variation as a foundation for investigating the relationship between genotype and phenotype. The Phase 3 of the project contains whole genome sequence data for 2,504 individuals from 26 populations. The columns (1KGP_AF, 1KGP_AFR_AF, 1KGP_AMR_AF, 1KGP_EAS_AF, 1KGP_SAS_AF) report the variant allele frequency for all individuals sequenced in phase 3 of the project (AF) and the variant allele frequency in the 5 main sub-populations of the project (AFR – African, AMR – American, EAS – East Asia, EUR – European, SAS – South Asia.)
- ESP6500 – The goal of the NHLBI GO Exome Sequencing Project (ESP) is to discover novel genes and mechanisms contributing to heart, lung and blood disorders by pioneering the application of next-generation sequencing of the protein coding regions of the human genome across diverse, richly-phenotyped populations and to share these datasets and findings with the scientific community. The current ESP6500 release contains whole exome sequence data for approximately 6500 individuals. The columns ESP6500 AA and ESP6500 EA report the variant allele frequency for all sequenced African American (AA) individuals and for all sequenced European American (EA) individuals.
- GnomAD – The Genome Aggregation Database (gnomAD) is a resource developed by an international coalition of investigators, with the goal of aggregating and harmonizing both exome and genome sequencing data from a wide variety of large-scale sequencing projects, and making summary data available for the wider scientific community. The columns report the variant allele frequency in all the genomes (gnomAD_maf) and in several sub-populations: African/African-American (gnomad_AFR_maf), Latino/Admixed American (gnomad_AMR_maf), Ashkenazi Jewish (gnomad_ASJ_maf), East-Asian (gnomad_EAS_maf), Finnish (gnomad_FIN_maf), Non-Finnish European (gnomad_NFE_maf), other (gnomad_OTH_maf), South Asian (gnomad_SAS_maf).
- Tool predictions – [Small variants only! Results of submitting the variant to some pathogenicity prediction tools. These predictions support the effect of the variant.
- SIFT – SIFT predicts whether an amino acid substitution affects protein function. A SIFT prediction is based on the degree of conservation of amino acid residues in sequence alignments derived from closely related sequences, collected through PSI-BLAST. SIFT can be applied to naturally occurring nonsynonymous polymorphisms or laboratory-induced missense mutations. Two different values can be found: 1) missense genetic variant is predicted as deleterious (D) or 2) is predicted as tolerated (T). You can find more information in the original publication: Kumar P et al. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4(7), 1073-1081 (2009). PMID:19561590
- PP2 HVAR – PolyPhen-2 (Polymorphism Phenotyping v2) is a software tool which predicts possible impact of amino acid substitutions on the structure and function of human proteins using straightforward physical and evolutionary comparative considerations. Three different values can be found: 1) missense genetic variant is predicted as probably Damaging (D), 2) is predicted as Possibly damaging (P) or 3) is predicted as Benign (B). Predictions are based on the HumVar dataset. You can find more information in the original publication: Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations POLYPHEN2. Nat Methods 7, 248–249 (2010). PMID: 20354512
- MutationTaster – The column reports the MutationTaster prediction for the corresponding variant. There are 4 possible values: ‘disease causing’ if the variant is probably deleterious, ‘disease causing automatic’ if the variant is known to be deleterious (due to its presence in Clinvar or HGMD), ‘polymorphism’ if the variant is probably harmless and ‘polymorphism automatic’ if the variant is known to be harmless (since it is present multiple times in the 1000 genomes project or HapMap). More details can be found in the original publication: Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014 Apr;11(4):361-2. PMID: 24681721
- CADD – CADD raw score for functional prediction of a SNP. The larger the score the more likely the SNP has damaging effect. Please note the following copyright statement for CADD: “CADD scores (http://cadd.gs.washington.edu/) are Copyright 2013 University of Washington and Hudson-Alpha Institute for Biotechnology (all rights reserved) but are freely available for all academic, non-commercial applications. For commercial licensing information contact Jennifer McCullar (mccullaj@uw.edu).”. Please refer to Kircher et al., Nature Genetics (2014) for more details.
- Gene Name – Name of the gene as according to the HUGO Gene Nomenclature Committee
- Chromosome – The chromosome on which the gene is located in humans.
- Uniprot Accession Number – The Universal Protein Resource (UniProt) is a comprehensive resource for protein sequence and annotation data. The Uniprot ACC represents the unique protein accession number for the gene-of-interest provided by UniProt
- Ensembl gene ID - Ensembl is a genome browser integrating different tools for gene annotation and variant prediction. The Ensembl gene ID represents the unique Ensembl identifier of the gene-of-interest in the Ensembl database.
- Entrez gene ID Entrez Gene is the database of the National Center for Biotechnology Information (NCBI) for gene-specific information. The Entrez gene ID represents the unique identifier of the gene-of-interest in the Entrez database.
- Gene Ontology Molecular Function – Gene Ontology (GO) terms for molecular function from UniProt.
- Pathways – The column reports the unique pathways IDs associated with the gene-of-interest in Reactome and KEGG. Reactome is a free, open-source, curated and peer reviewed pathway database. Kyoto Encyclopedia of Genes and Genomes (KEGG) is a database resource for understanding high-level functions and utilities of the biological system, such as the cell, the organism and the ecosystem, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies.
- Essential In Mouse – Known essential status of the gene. Two different values can be found: 1) essential (E) or 2) non-essential phenotype-changing (N) based on the Mouse Genome Informatics database. You can find more information in the original publication: Georgi, B., Voight, B. F. & Bućan, M. From Mouse to Human: Evolutionary Genomics Analysis of Human Orthologs of Essential Genes. PLoS Genet 9, (2013). PMID:23675308
- Variants – Genetic variants for this gene that are present in OLIDA.
- Combinations – Combinations in which variants of this gene are involved.
- Diseases – Diseases in OLIDA which are linked to combinations in which the gene-of-interest is involved.
- Genes - Names of the genes that make up this combination.
- Gene relationship - Type of relationship between the genes involved carrying the mutations. This information was obtained as the consensus between the different articles that identified the combinations, articles referenced by the authors of these articles and external databases specified for each type of interaction. When the combinations involve more than two genes, an interaction term is listed if it represents the interaction of at least two genes but is not necessarily applicable to all pairs of genes in the combination. 10 types of relationships were identified:
- Direct interaction - there is a direct protein-protein interaction between the protein products of the genes. The external database information was retrieved from comPPI.
- Indirect interaction - there is an indirect or “two step” interaction between the protein products of the two genes. If protein “A” and protein “B” interact with protein “C”, protein A and protein B are indirectly interacting. In other words, they share a common interactor.
- Same protein complex - the protein products of the genes are not necessarily known to be directly interacting, but they are part of the same protein complex.
- Same pathway - the genes are involved in the same biological pathway. The external database information was retrieved from KEGG and Reactome.
- Relevant pathways for phenotype - the genes are involved in different biological pathways, but these pathways are relevant to the phenotype of interest. The external database information was retrieved from KEGG and Reactome and the pathways were then checked manually to be relevant to the phenotype.
- Co-expression - the expression of the mRNA products of the genes are correlated.
- Similar function - the protein products from both genes contain the same functional conserved motifs or conserved domains.
- Involved in the same disease - The genes are known to be associated with the same disease.
- Affecting the same tissue - the genes are expressed in at least one common tissue or organ.
- Same cellular compartment - the protein products of the genes are located in the same cellular compartment.
- Protein interactions - This column contains the list of pairs of genes involved in the combination that are directly interacting according to comPPI.
- Common Pathways - List of pathways in which at least two of the genes that make up the combination are involved.
- GENEmeta - The score of the evidence supporting the relationship between the genes and their relevance to the phenotype of interest, based on both article information and external databases.
- OLIDA combinations IDs - OLIDA identifiers of the combinations that contain variants in these genes.
- Disease name (ORPHANET) – Orphanet is the reference portal for information on rare diseases and orphan drugs, for all audiences. Orphanet’s aim is to help improve the diagnosis, care and treatment of patients with rare diseases. This column represents the name of the disease as present in Orphanet.
- ORPHANET ID – The column reports the identifier for the disease as retrieved from Orphanet.
- ICD-10 ID – The International Classification of Diseases (ICD) is the standard diagnostic tool for epidemiology, health management and clinical purposes. ICD is used to classify diseases and other health problems recorded on many types of health and vital records, including death certificates and health records. This column reports the ICD-10 disease identifier as obtained from the ICD-10 online version: 2015.
- ICD-10 Category – The column reports the ICD-10 chapter disease category as obtained from the ICD-10 online version: 2015.
- OMIM ID – The Online Mendelian Inheritance in Man (OMIM) is an online catalog of human genes and genetic disorders. The column reports the OMIM disease identifiers linked to the disease-of-interest as present in Orphanet.
- Combinations – The oligogenic variant combinations to which the disease-of-interest is linked.
API Documentation
In addition to the web interface for this website, a REST API is also provided for machine readable access of the data. The contents of the API can be explored through the API page. The API allows for retrieval of data with HTTP GET requests to the available endpoints with a token authentification. An OPTIONS HTTP request will return metadata that describes the API endpoints. Documentation on how to use this API is available as a Swagger-UI or as a Redoc interface. The schema of the API follows the OpenAPI standard and is formatted according to the JSONAPI specification. The specification for the API is available in both JSON and YAML formats.
Authentication
A token can be obtained through POST request with the command :
curl -X POST "https://olida.ibsquare.be/api-token/" -d "{ \"username\": \"user\", \"password\": \"yourpassword\"}"
with user and yourpassword replaced with your credentials. The API should respond in a similar way as follows:
{"token": "9944b09199c62bcf9418ad846dd0e4bbdfc6ee4b"}
The token can then be used in the header for all the requests sent to the API as in the example below:
curl -X GET https://olida.ibsquare.be/api/combinations/ -H 'Authorization: Token 9944b09199c62bcf9418ad846dd0e4bbdfc6ee4b'
Authentication to the API can also be done through the Swagger and Redoc interfaces by login to the Django session.
This API was created using the Django REST framework extension.
Tutorials
Click the headers below to expand tutorials on those subjects
All curated data from OLIDA can be accessed from the Browse page, which is shown in Figure 1.1.
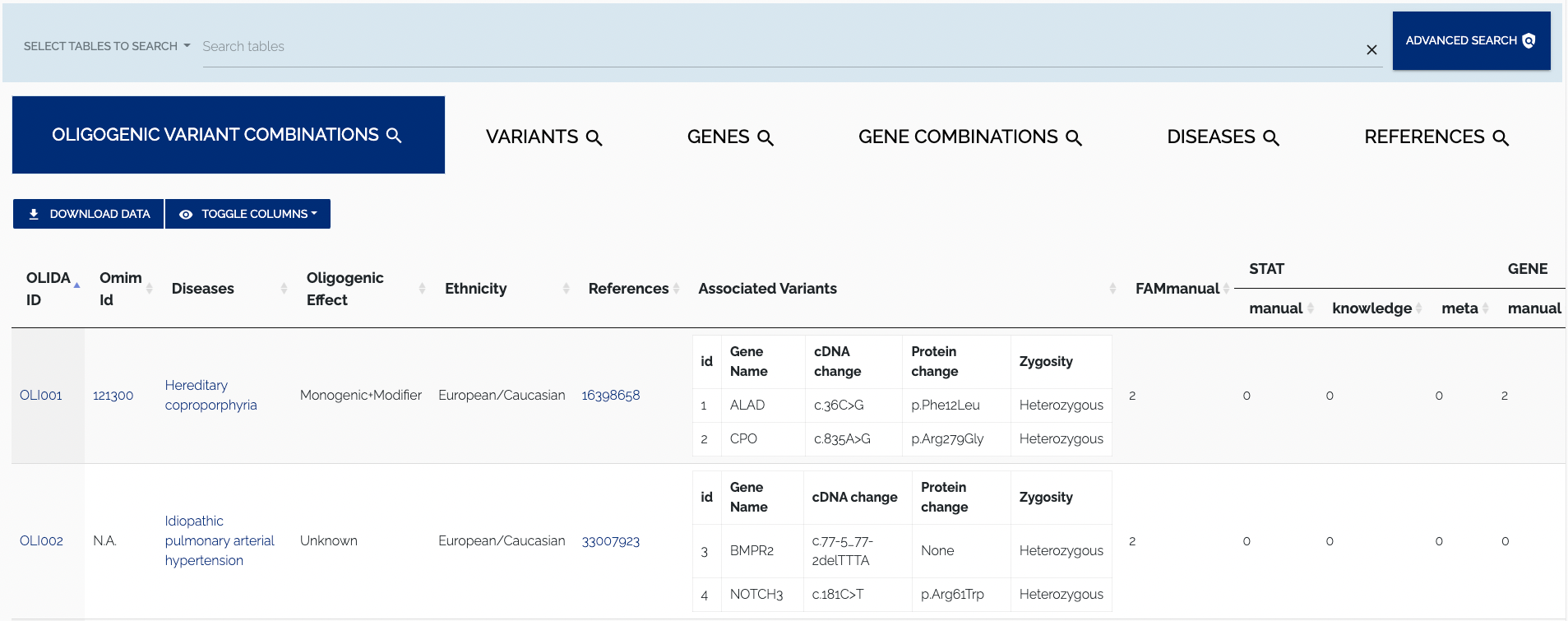
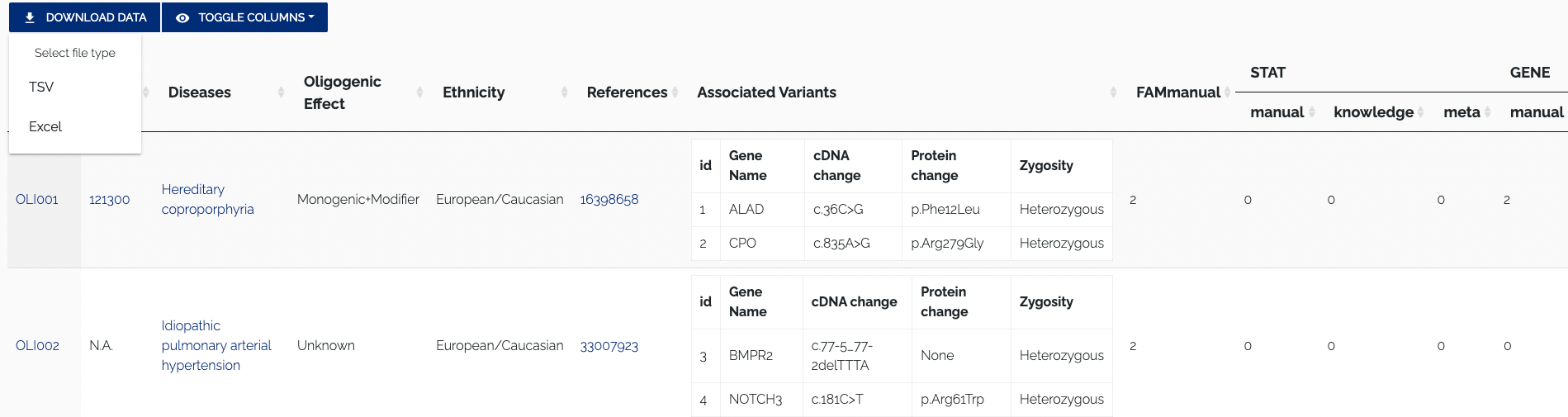
For every table, the top left contains buttons to download data and toggle columns on or off in the displayed table. The "Download" button allows for the data to be downloaded by selecting one of the supported formats from the dropdown menu. Currently, the TSV and Excel formats are supported. Figure 1.2 below shows a closeup of the format selection dropdown for downloading data.
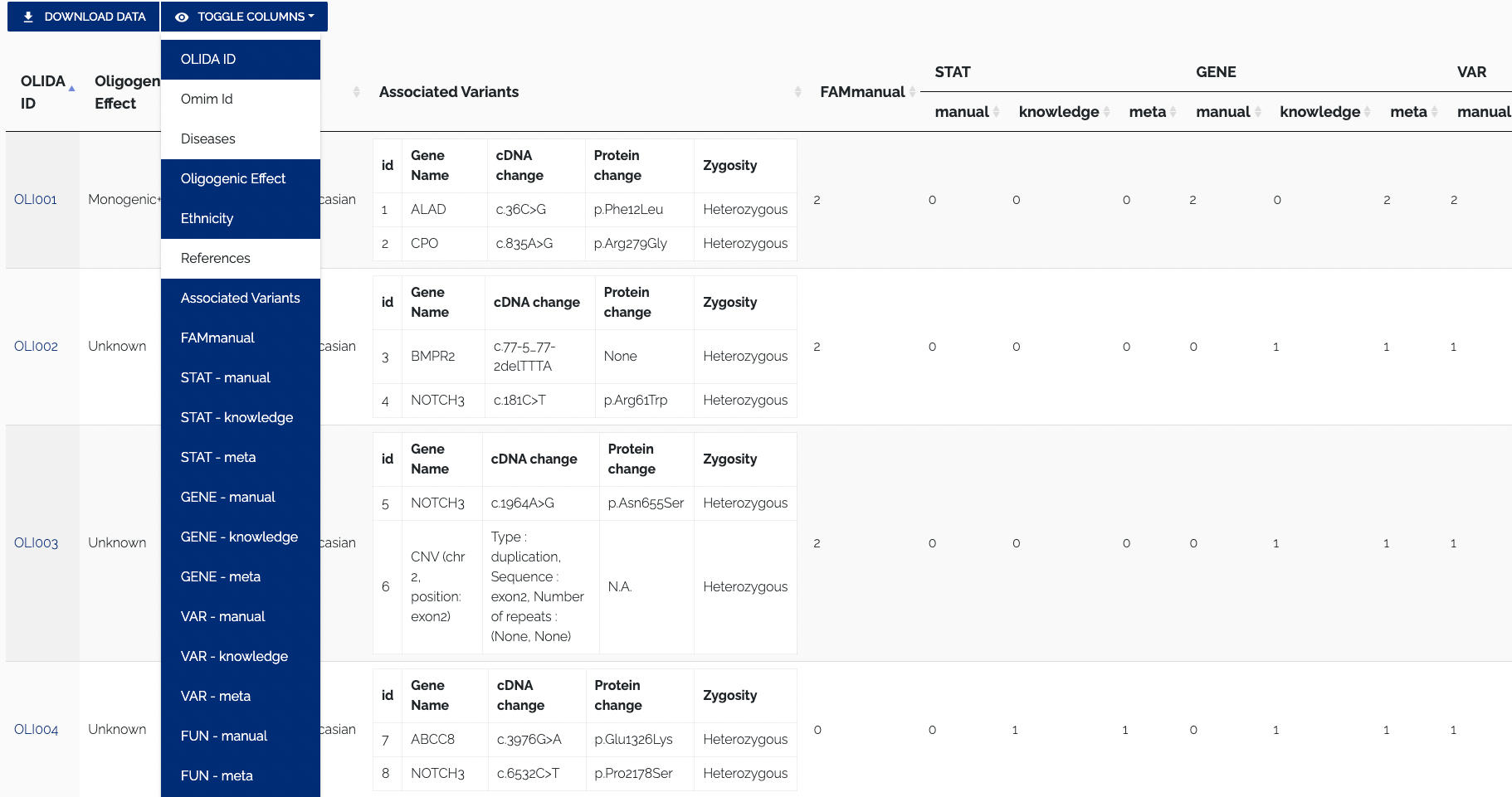
To download the data in JSON format, one can use the REST API. To toggle columns of the table, simply click the column visibility dropdown and select or unselect a column name. By default all columns of a table are shown, columns that do not fit on the screen are displayed to the right and can be viewed by scrolling horizontally. Names of active columns will have a blue background, while the name of hidden columns will have a whit background. Figure 1.3 below shows the Oligogenic variant Combinations table with some columns disabled.
To search for a particular entry in the tables that are displayed in the Browse page, the main search bar on top of the page can be used. Users can select which tables that should be searched by enabling/disabling the elements in the dropdown to the left of the search input. By default all tables are searched. To disable search for a particular table one has to click on the corresponding element in the dropdown. Clicking on a disabled item will re-enable search for the associated table. Additionally, one can Ctrl/Cmd+click an element to search the selected table only and disable search for all others. Only visible columns will be searched. Figure 1.4 shows an example of search with certain tables being excluded.

The Advanced search feature is still under development. A tutorial on how to use it will be added here when it is ready.
Besides browsing through the existing data, we also allow users to submit their research data in order to expand the contents of the database. New submissions will only be accepted after careful manual inspection to ensure that all provided data is of high quality. The submission wizard is available from the SUBMIT page. Note that we require submitters to either be logged in with a user account or to minimally provide basic information that will allow us to contact the user.
Overview of the submission wizard steps:
- Identity: If you are already logged in this step will automatically be skipped. Otherwise one will be asked to log in or at least input a valid e-mail address so we can contact you for potential additional information or if a submission was accepted or rejected.
- Publication: In this step we minimally require a user to submit a PubMed ID or DOI of the publication that describes the new data. The server will then automatically try to retrieve the necessary information on the publication and display it in a filled in form. The user can then update their data if necessary and provide additional information if they wish so.
- Combination:
Select the oligogenic variant combination(s) that are described by this new publication.
It is also possible to submit new combinations. Creating a new combination will take you
through the following steps:
- Associate variants
- Associate disease(s)
- Description of the combination properties
- Verification: The final step in the wizard will display hte new and modified data that would be added or updated from this submission. The user can then go back to fix mistakes or validate the submission after which it will be curated.
Detailed description of wizard steps
The slides below show the different steps of the submission wizard and detail what data should be provided for each step. Below each slide a description of the step is provided and the data or actions which are required or optional in the step are discussed.
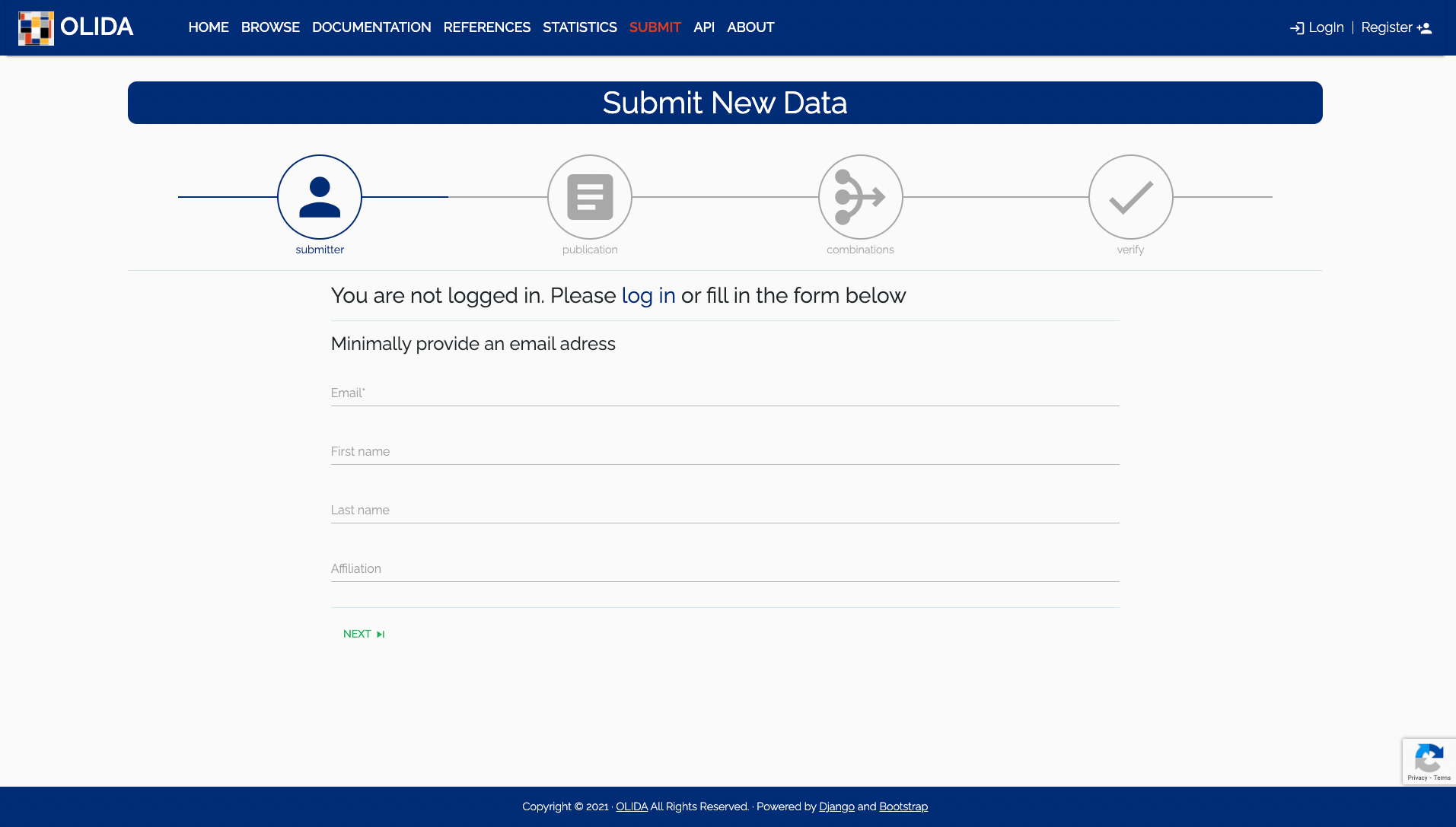
Submitter Info
Minimally provide an email address and optionally a name and affiliation.
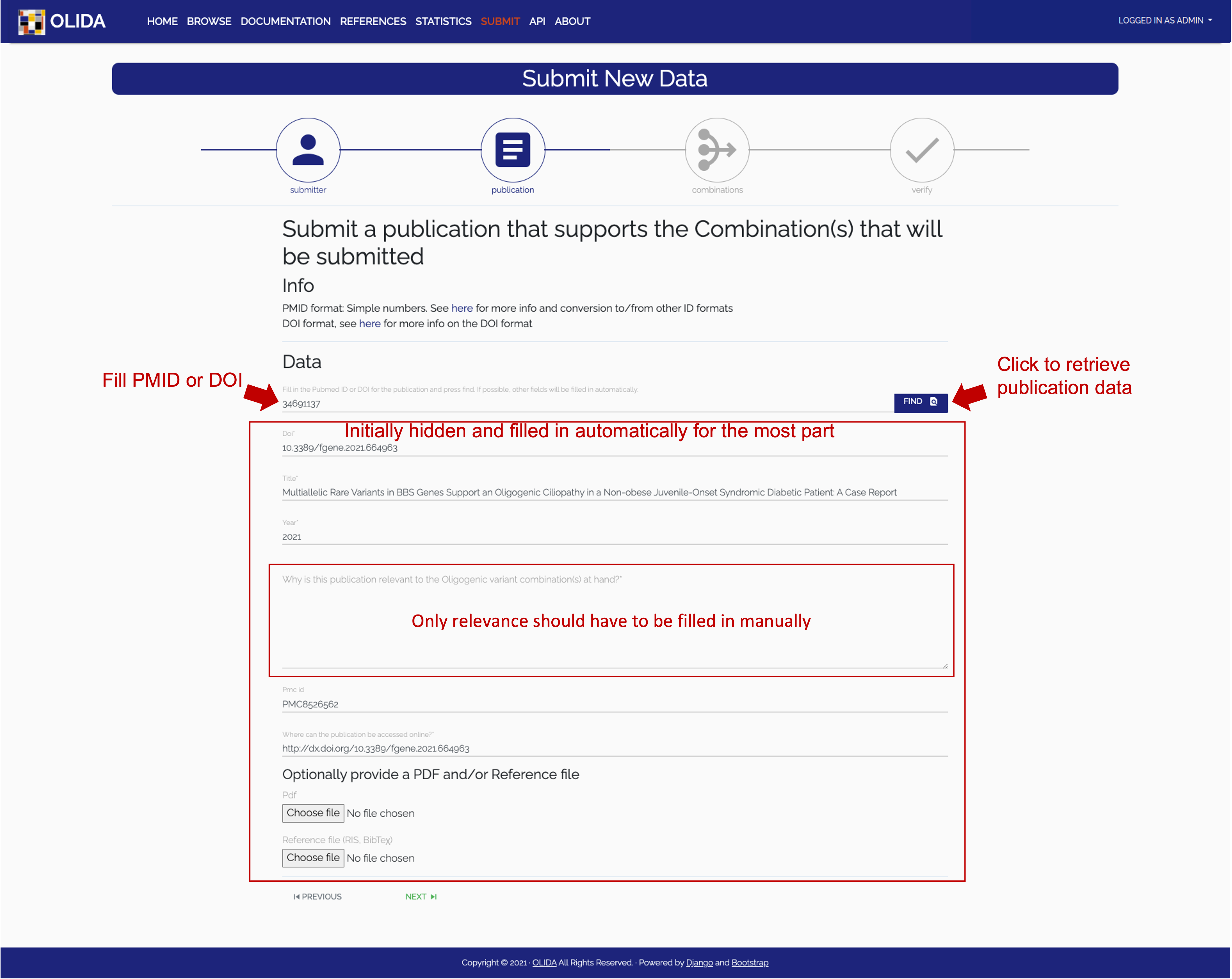
Data on the Publication
Required and optional information regarding the publication that discusses the new data to be submitted.
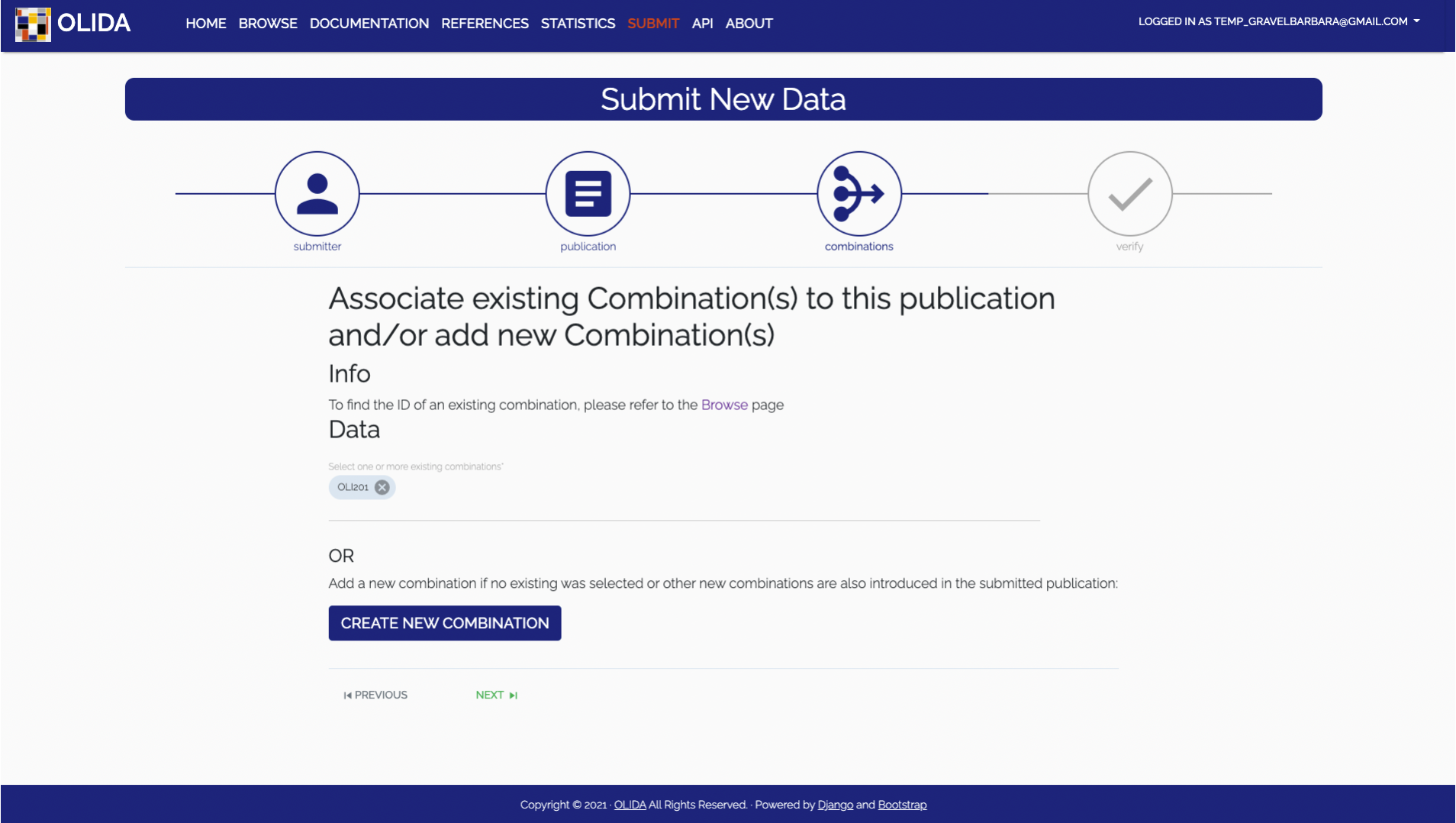
Combination Selection and/or Creation
Select existing combinations that are discussed in the previously submitted publication. New combinations can also be created from this page and will automatically be added to the selection.
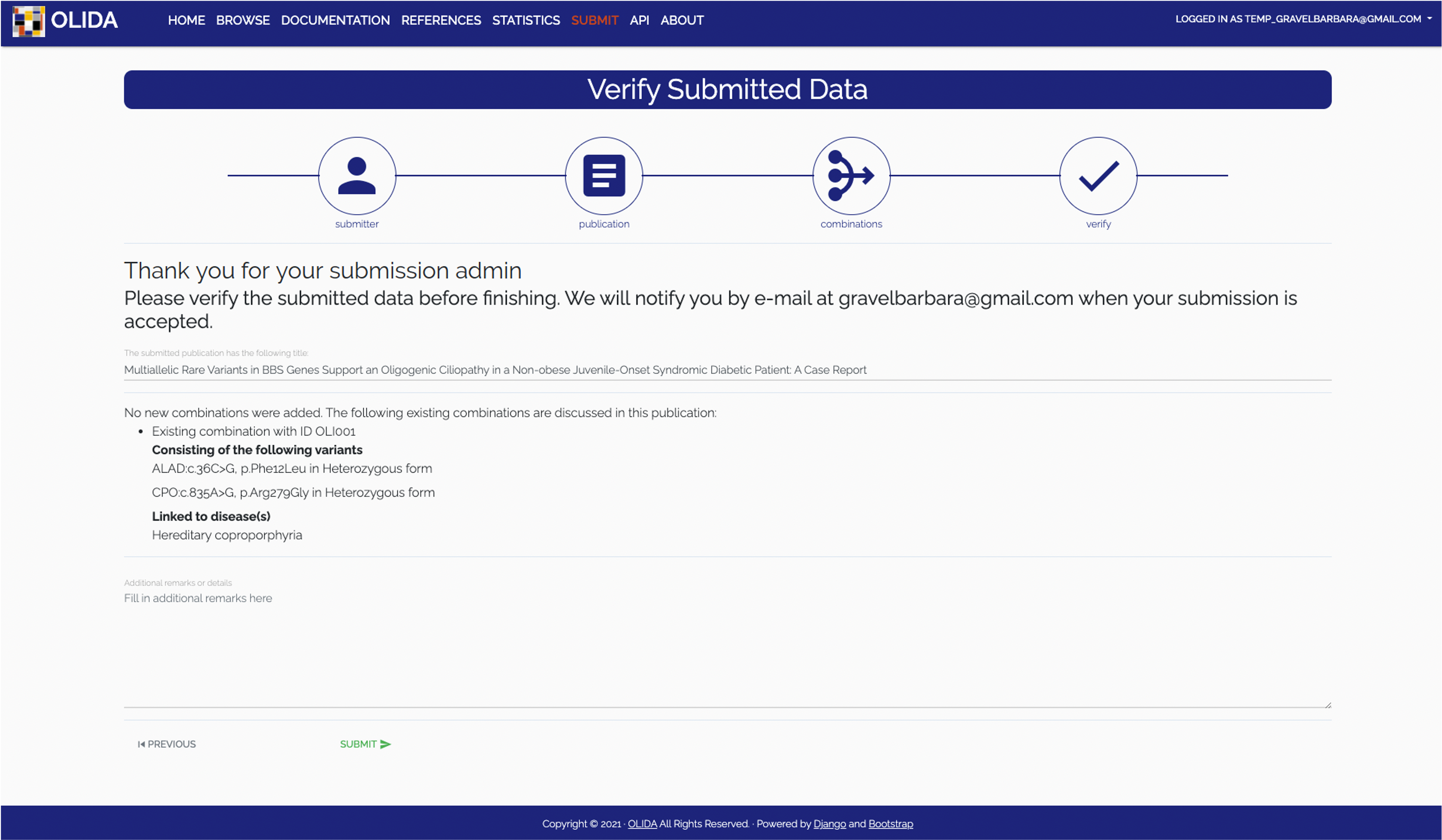
Verify Submitted Data
Verify that the submitted data is correct and leave remarks.
- Required: To guarantee the quality of submitted data and to be able to contact a submitter if additional information is required, at minimum an email address is requested.
- Optional: Optionally, a name (first and last) and an affiliation is useful in identifying the submitting person and give support to the quality of the submitted data. Logged in users will start at the next step and not see this step in the wizard progress bar.
- Required: Submitters should verify that the retrieved data is correct and properly formatted (e.g. in the currently shown slide, the abstract entry contains tags that should not be submitted). The submitter should also fill in the relevance field indicated on the slide with a free-text description of why this publication and the new data it discusses is relevant to be added to the OLIDA dataset.
- Optional: Optional files can be uploaded, being a PDF of the publication text or a bibliography file (BibteΧ or RIS) for easy referencing.
- Required: The user has the choice to either select an existing combination already present in the database or to create a new combination. To select an existing combination, one will be presented with an auto filled input which will propose matching combinations based on the OLIDA id. To know the OLIDA ID of a combination, one can consult the Browse page. If the combination that is discussed in the publication can not be found in the current dataset, one can click the CREATE COMBINATION button, which will open a modal with a new form wizard for creating combinations.
- Optional: No optional data can be provided in this step.
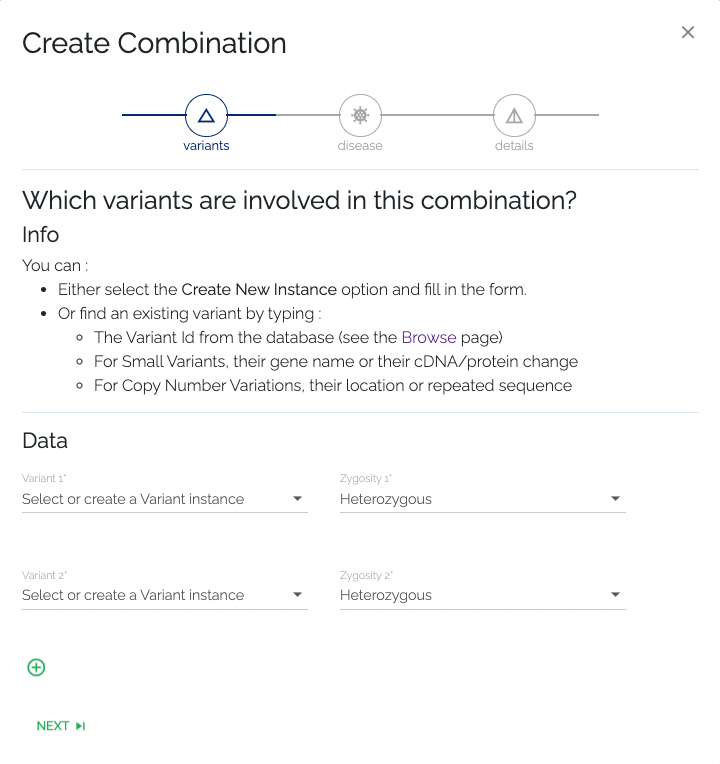
- Required: To associate a variant, one can select an existing variant by clicking the dropdown element and selecting from the list, which can be filtered by typing in the input field. Variants can be filtered by their gene name, cDNA or protein change, or by the ID that can be found on the Browse page. Alternatively, a new variant can be added to the database by selecting the Create new instance option from the dropdown menu that is used to select existing variants. After selecting this option, the user is presented with a dropdown where they should select the type of variant they want to add. After the user selects the type of variant they wish to submit, they will be presented with a form that requests the information that is necessary to add the selected type of variant. When the form is submitted successfully, the new variant will be added to the database and selected automatically. One has to select or create at least two variants in this step.
- Optional: If the new combination consists of more than two variants, one can add variants by clicking the + button at the bottom of the form. While this action is optional, it is left up to the submitter to know how many variants are required to form a given combination.
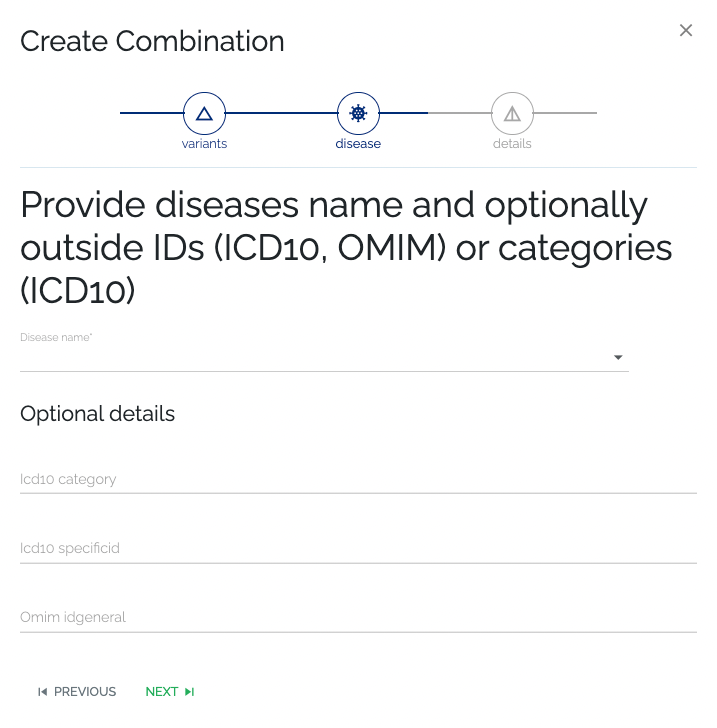
- Required: As with previous steps, the user can select an existing disease or create a new one by typing in the name and going to the next step. If a new disease name was given, an attempt will be made to automatically retrieve additional information on the disease.
- Optional: Additional information can be provided by the submitter, by filling in the optional fields below the disease name selection. The information that was provided by the submitter will take precedence over automatically retrieved data.
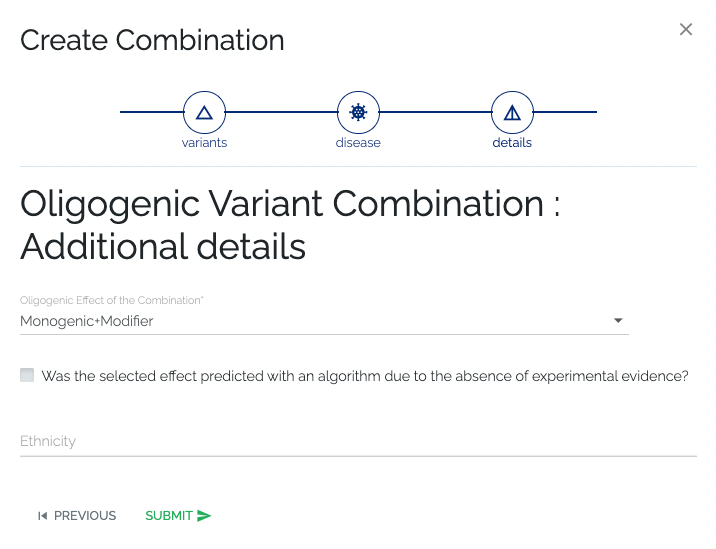
- Required: No information is required.
- Optional: Optional details such as the oligogenic effect and if it was predicted, the ethnicity of the patient carrying the combination can be provided.
The API can be used directly through the GUI as a Swagger-UI or as a Redoc interface.
However, you can also access to the API programmatically as long as you possess a username and the related password to OLIDA. You can find a Python script example here.
Frequently Asked Questions
If you still have questions after reading this documentation, you can contact us at olida@ibsquare.be. We will try to get in touch with you as quickly as possible and if relevant will add the question and answer to this F.A.Q..